
Pathologie moléculaire/cytosquelette
Pathologie moléculaire du cytosquelette
La définition des constituants du cytosquelette est expérimentale. Il s’agit de l’ensemble des éléments filamenteux du cytoplasme des cellules eucaryotiques qui persistent après un traitement des cellules avec un détergent doux qui élimine les constituants membranaires et les composants solubles du cytoplasme.
Cela comporte :
- les filaments intermédiaires,
- les microfilaments ou filaments d’actine,
- les microtubules et le réseau microtrabéculaire.
Par ces cycles continus assemblage-déassemblage, le cytosquelette participe à d’importantes fonctions de la cellule comme la motilité, le maintien de la forme cellulaire, la croissance et la division cellulaire, la sécrétion, l’adhésion, la phagocytose et le contact cellule-cellule.
Le cytosquelette établit également un lien entre le cytosquelette et l’enveloppe nucléaire. Par ailleurs, à travers le cytosquelette, les cellules peuvent influencer les cellules environnantes par le biais des jonctions intercellulaires ou des effets sur la matrice extracellulaire.
Le cytosquelette est constitué de trois familles de protéines fibrillaires : *les filaments intermédiaires,
- les microfilaments
- les microtubules.
Les maladies causées par les anomalies moléculaires du cytosquelette constituent le champ en expansion rapide des cytosquelettopathies.
Pathologie des filaments intermédiaires
[modifier | modifier le wikicode]Les filaments intermédiaires doivent leur nom à leur taille, qui est intermédiaire entre celles des microfilaments et des microtubules.
Les filaments intermédiaires constituent une famille hautement diversifiée de protéines fibrillaires, présentes chez tous les métazoaires, qui s’assemblent pour former des filaments de 10 nm d’épaisseur dans le cytoplasme et le noyau. Ils sont composés de 8 sous-filaments, qui s’assemblent pour former une fibre. Tous les filaments intermédiaires possèdent 3 régions : tête, corps et queue.
Ils différent des microfilaments d’actine et des microtubules de tubuline par leur nombre important, leur distribution cytoplasmique et nucléaire, leurs structures primaires, leur architecture non-polaire, leur relative insolubilité et leur dynamique indépendante des nucléotides.
Leur fonction principale est de maintenir la forme cellulaire en tissant un réseau entre les différents composants cellulaires. Pour cela, les protéines des filaments intermédiaires interagissent des protéines très diverses qui peuvent se diviser en lieuses, assembleuses, chaperones, kinases, protéines liées à l’apoptose et protéines nucléaires.
En plus de leur rôle architectural, les filaments intermédiaires protègent contre le stress non-mécanique et diminuent la susceptibilité à l’apoptose.
D’autres fonctions sont spécifiques de tissus; ainsi, les neurofilaments jouent un rôle dans l’arborisation dendritique et dans la croissance radiaire des axones myélinisés.
Le génome humain contient au moins 65 gènes fonctionnels codant les protéines des filaments intermédiaires, ce qui en fait une des 100 familles de gènes les plus importants.
Les filaments intermédiaires forment une famille complexe divisée 5 groupes :
- Types I and II: les kératines
- Type III : la desmin, la vimentin, la GFAP, la périphérine
- Type IV: les neurofilaments
- Type V: les lamines
- Type VI: la nestine
Un aspect important des filaments intermédiaires est leur expression selective dans certains types cellulaires et pendant la différenciation. Cela explique que les mutations de ces gènes entraînent des maladies spécifiques d’organes ou des tissus. Ainsi, les kératines (KRTs) s’expriment dans les cellules épithéliales, la vimentine dans les cellules mésenchymateuses, la desmine dans les cellules musculaires, les filaments gliaux (GFAP) dans les astrocytes et les neurofilaments dans les neurones.
Du fait de cette spécificité d’expression, l’étude immunohistochimique des protéines des filaments intermédiaires dans les tumeurs peu différenciées permet de suggérer une différenciation tumorale. Par exemple, l’immunodétection des kératines permet de définir une tumeur épithéliale, la desmine, une tumeur musculaire. Ces études permettent également de détecter des micrométastases.
Plus de 30 maladies liées à des mutations des gènes codant les filaments intermédiaires ont été décrites. Les effets de ces mutations dépendent de la nature des protéines et de leur localisation cellulaire. Les filaments intermédiaires défectifs peuvent mal se polymériser, se désorganiser et entrainer des pathologies sévères.
La perte de l’intégrité cellulaire et nucléaire après une exposition à des traumatismes physiques (par exemple, pression, étirement et chaleur) est une marque des anomalies de plusieurs types de filaments intermédiaires et indique que la fragilité cellulaire est un facteur majeur de la pathogénie de ces anomalies.
Des mutations peuvent diminuer ou augmenter les interactions entre les filaments intermédiaires et les organelles ou les granules. Elles peuvent également perturber le trafic intracellulaire des protéines, ce qui entraîne des anomalies des fonctions cellulaires et une plus grande susceptibilité à l’apoptose. Par exemple, les mutations proline to leucine en position 24 de la kératine-5 (KRT5) entraîne une épidermolyse bulleuse simple avec anomalies de la pigmentation en association avec une distribution aberrante des mitochondries et des granules de mélanine; les souris dont le gène de la kératine-8 est inactivée ont un mauvais adressage subcellulaire des protéines dans les hépatocytes et les cellules intestinales; les souris dont la desmine est inactivée ont une distribution anormale des mitochondries.
Les dysfonctionnements et la mort cellulaire peut aussi survenir pat des mutations entraînant un gain de fonction pour la protéine mutée. Ainsi, les agrégats cytoplasmiques et les inclusions observées dans les mutations de la desmine, de la GFAP (glial fibrillary acidic protein) et de plusieurs kératines reflètent l’inabilité de la cellule à gérer les protéines mutantes des filaments intermédiaires et entraîne des dysfonctions cellulaires pouvant conduire à la mort de la cellule. Ainsi, les lignées cellulaires issues de patients ayant une épidermolyse bulleuse par mutation des gènes codant la kératine-5 (KRT5) et la kératine-14 (KRT14) ont une sensibilité accrue au stress osmotique et une augmentation de l’activité des kinases de stress.
Les cytokeratines (cytokératinopathies)
[modifier | modifier le wikicode]Les kératines (KRTs) forment le plus important groupe de protéines des filaments intermédiaires. Elles sont les protéines les plus abondantes dans les cellules épithéliales.
Leur fonction primaire est de protéger les cellules épithéliales contre les contraintes mécaniques et non-mécaniques qui peuvent résulter en la mort cellulaire. D’autres fonctions des kératines sont la signalisation cellulaire, la réponse au stress et l’apoptose.
Les protéines de kératine subissent une régulation complexe associant des modifications post-traductionnelles et des interactions entre elles et avec différentes classes de protéines associées.
Aujourd’hui, environ 20 polypetides différents peuvent être distingués. Deux sous-types ont été définis selon des homologies de séquence :
- les kératines de type I (KRT9 à KRT20) sont plus petites (40-56.5 kDa) et relativement acides
- les kératines de type II (KRT1 à KRT8) sont plus grandes (53-67 kDa) et relativement basiques ou neutres. Leurs gènes sont situés sur le chromosome 12.
Les kératines constituent un réseau très dynamique de filaments cytoplasmiques de 10 à 12 nm, constitués d’hétéropolymères de chaîne de type I et II selon un rapport môlaire 1:1.
Certains sous-types de kératines s’expriment uniquement dans certains tissus. Ainsi, les epithelia simples expriment les kératines 7, 18, 19, et 20, alors que les epithelia complexes expriment les kératines 5/6, 10, 14, et 15.
Galerie
[modifier | modifier le wikicode]-
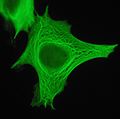 Cytokeratin filaments
Cytokeratin filaments -
 CK3 and CK12 in Corneal epithelium
CK3 and CK12 in Corneal epithelium -
 Cytokeratin 8
Cytokeratin 8 -
 Immunohistochemistry of small intestinal adenocarcinoma with cytokeratin 7, 19 and 20
Immunohistochemistry of small intestinal adenocarcinoma with cytokeratin 7, 19 and 20



Pathologie des kératines (Kératinopathies)
[modifier | modifier le wikicode]Un grand nombre de maladies sont associées à des mutations, en général dominant négatives, des gèenes de kératine. La plupart de ces mutations sont des mutations faux-sens ou de petites insertions ou des délétions décalant le cadre de lecture, et touchant principalement le corps de la molécule de kératine. Les mutations touchant les motifs hélice frontière du corps entraîne en général une maladie sévère et ont une forte pénétrance.
Les mutations ayant lieu ailleurs causent une maladie moins sévère ou une simple prédisposition, comme pour les gènes codant les kératine-8 (KRT8) et -18 (KRT18).
Les mutations moins actives peuvent conduire à des maladies touchant seulement les zones cutanées les plus exposées à la pression comme les paumes et les plantes, comme dans les hyperkératoses épidermolytiques palmo-plantaires.
| Kératine | Abbr. | Maladie |
|---|---|---|
| keratin 1 | KRT1 | |
| keratin 2 | KRT2 | |
| keratin 3 | KRT3 | |
| keratin 4 | KRT4 | |
| keratin 5 | KRT5 | |
| keratin 6 | KRT6 | |
| keratin 7 | KRT7 | |
| keratin 8 | ||
| keratin 9 | KRT9 | |
| keratin 10 | KRT10 | |
| keratin 11 | KRT11 | |
| keratin 12 | KRT12 | |
| keratin 13 | KRT13 | |
| keratin 14 | KRT14 | |
| keratin 15 | KRT15 |
Pathologie des kératines conventionnelles
[modifier | modifier le wikicode]Les études de microscopie électronique (ultrastructure) réalisées dans les années 80 ont montré que les cellules basales épidermiques des patients ayant une épidermolyse bulleuse simplex contiennent des agrégats denses cytoplasmiques.
Entre les années 80 et 90, diverses expérimentations ont montré que les mutations des gènes de kératines provoquait une agrégation des kératines dans le cytoplasme des cellules cultivées et une fragilité mécanique des cellules épithéliales in vivo.
Peu après, des mutations des gènes codant la kératine-5 (KRT5) et la kératine-14 (KRT14) ont été décrites chez des patients ayant une épidermolyse bulleuse simplex. Chez ces patients, on observe la formation de bulles après un traumatisme frictionnel dans la zone basale de l’épiderme, où la kératine-5 (KRT5) et la kératine-14 (KRT14) sont les plus abondantes.
Les mutations dominantes des gènes codant la kératine-1 (KRT1) ou son partenaire, la kératine-10 (KRT10), s’exprimant dans l’épiderme suprabasal, sont à l’origine d’hyperkératose épidermolytique.
Les maladies des kératines sont caractérisées par une fragilité, ou parfois une hyperplasie, des tissus exprimant la kératine. Les filaments périnucléaires se rétractent dans le cytoplasme ce qui entraîne une baisse de la résistance cytoplasmique aux déformations cellulaires.
Les mutations qui altérent les modifications post-traductionnelles ou les interactions avec les autres protéines cellulaires peuvent aussi entraîner une fragilité cellualire en perturbant les propriétés et la fonction des filaments intermédiaires.
Les mutations de la kératine-8 (KRT8) et de la kératine-18 (KRT18) diminuent la solubilité de ces keratins, perturbent leur phosphorylation et leur glycosylation. Les mutations de ces deux gènes qui permettent la formation du filament sont un facteur de risque pour la cirrhose hépatique. De plus, certaines mutations de KRT8 seraient des facteurs de prédisposition aux maladies inflammatoires du tube digestif et à la pancréatite chronique.
Les mutations de la kératine-14 (KRT14) altèrent la dynamique cytosquelettique et la solubilité de la protéine lorsque le gene mute est introduit dans les cellules épithéliales.
Pathologie des kératines des poils et des cheveux
[modifier | modifier le wikicode]Le monilethrix est une kératinopathie caractérisé par une irrégularité et une fragilité des cheveux. Il est causé par des mutations des gènes KRTHB1 et KRTHB6, codant respectivement les kératines pilaires keratins Hb1 and Hb6.
Plusieurs autres kératines épithéliales s’expriment spécifiquement dans les follicules pilaires, comme la kératine-6hf. Des variations de séquence du gène de cette protéine sont des facteurs de risque du syndrome loose-anagen et de la pseudo-folliculite de la barbe.
Le chevauchement de la distribution des kératines dans les follicules pilaires expliquent probablement la rareté des kératinopathies touchant les cheveux.
Cependant, il est probable que les gènes de kératine jouent un rôle dans genèse de plusieurs maladies des cheveux ou dans des variations normales de leurs caractéristiques.
Pathologie des desmines (desminopathies)
[modifier | modifier le wikicode]La desmine est un filament intermédiaire de type III, spécifique du muscle, exprimé dans le muscle lisse et le muscle strié cardiaque et squelettique.
Les desminopathies constituent un groupe de maladies identifié en 1998 et causé par des anomalies génétiques du gène codant la desmine. Elles entraînent une dystrophie musculaire, la myopathie avec surcharge en desmine.
alpha B-crystallin (CRYAB)
[modifier | modifier le wikicode]Les mutations gain-de-fonction du gene CRYAB codant l’alpha B-crystalline sont également à l’origine d’une desninopathie. En effet, l’alpha B-crystalline est une protéine chaperone qui régule l’organisation de plusieurs filaments intermédiaires cytoplasmiques, comme la desmine dans le muscle et la phakinine et la filensine dans l’œil. Les mutations inactivatrices de CRYAB sont à l’origine de cataracte congénitale.
Pathologie des microfilaments "beaded"
[modifier | modifier le wikicode]Plus de 99% du cristallin, la lentille oculaire des vertébrés, est composé de cellules lenticulaires hautement spécialisées. Comme pour les globules rouges, cette différentiation inclut la perte de toutes les organelles cytoplasmiques membraneuses. Les cellules lenticulaires ne sont pas détruites après leur mort et persistent localement pendant toute la vie de l’organisme. Ce phénomène s’accompagne d’intenses remaniements du cytosquelette cellulaire. Bien que des microfilaments d’actine, les microtubules et les filaments intermédiaires de vimentine s’observent dans les cellules nouvellement formés, ils disparaissent précocement au cours de la différentiation.
Les deux protéines de filaments intermédiaires de la cellule lenticulaire sont la phakinine (CP49) et la filensine (CP115), qui s’expriment dès que la différentiation a commencé. Ces deux protéines constituent les éléments principaux d’une composante du cytosquelette appelée ‘filaments perlés’, très abondants dans les cellules lenticulaires du cristallin..
Les mutations de la phakinine sont à l’origine de cataracte dominante congénitale ou infantile. Il est assez probable que les mutations de son partenaire protéique, la filensine, puissent également donner le même tableau clinique.
Les mutations inactivatrices du gene CRYAB codant la protéine alpha B-crystalline, chaperone des protéines phakinine et filensine, désorganisent les filaments perlés et sont à l’origine de cataracte congénitale.
GFAP
[modifier | modifier le wikicode]La protéine fibrillaire acide gliale (GFAP pour glial fibrillary acidic protein) forme les fibres de Rosenthal dans plusieurs situations pathologiques comme les astrocytomes pilocytiques.
Les mutations germinales du gène de la GFAP sont à l’origine de la maladie d’Alexander, une maladie neurodégénérative.
Periphérine
[modifier | modifier le wikicode]La périphérine forme, comme la desmine, des filaments intermédiaires de type III. Cette protéine cytosquelettique s’observe dans les neurones du système nerveux périphérique et dans les neuroblastes en culture.
Des variants (polymorphismes) du gène de la périphérine entraînent des susceptibilités à la sclérose latérale amyotrophique (MIM.105400).
Les neurofilaments
[modifier | modifier le wikicode]Les neurofilaments sont des filaments intermédiaires spécifiques des neurones. Ils s’assemblent en s’associant par leur partie centrale, des domaines alpha-hélice de type coiled-coil. Les chaînes de neurofilaments sont classées selon leur masse moléculaire : chaîne lourde (NEFH), chaîne moyenne (NEFM) et chaîne légère (NEFL).
Les NEFH et les NEFM se différencient des NEFL, car elles contiennent un domaine carboxy terminal (queue) et forment des connexions avec les structures adjacentes et les autres neurofilaments. Les neurofilaments contiennent des motifs répétés KSP qui sont des motifs consensus pour les proline kinases, hautement phosphorylés in vivo. Leur fonction est donc certainement régulée par leur phosphorylation.
En association avec les autres composantes axonales comme les microtubules, ils forment le cytosquelette dynamique de l’axone. Ils gèrent et régulent la plasticité du cytosquelette axonal par la régulation de la croissance des neurites, du calibre axonal et du transport axonal. Pathologie (les neurofilamentopathies) : Les mutations de la chaîne légère de neurofilaments (NEFL) sont observées dans certaines formes de maladie de Charcot-Marie-Tooth axonale.
Les mutations de la chaîne lourde de neurofilaments (NEFH) sont des causes potentielles de sclérose latérale amyotrophique sporadique (MIM.105400). Cette pathologie peut également être causée par des mutations de la périphérine, un filament intermédiaire de type III présent dans les neurones des cellules du système nerveux périphérique et dans les neurones en culture.
Laminopathies
[modifier | modifier le wikicode]Les lamines constituent une famille de polypetides composant la couche protéinacée appose à la membrane interne nucléaire. Chez les mammifères, 3 lamines, lamine-A (LMNA), lamine-B (LMNB), et lamine-C (LMNAC), ont été décrites avec des poids moléculaires allant de 60,000 à 78,000. Les lamines –A et –C possédent une forte homologie de séquence et diffèrent de la lamine-C. Pathologie des lamines (Laminopathies)
Les laminopathies regroupent les maladies causées par les anomalies des protéines de l’enveloppe nucléaire. La plupart intéressent la lamine-A, qui s’exprime sur la plupart des cellules adultes.
Les laminopathies constituent un groupe de maladies génétiques exprimant un phénotype variable, causé par des mutations du gène LMNA qui code les isoformes A et C (lamine A/C), exprimées différentiellement lors du développement.
Les mutations du gêne LMNA ont été impliqués dans une dizaine de maladies. Il s’agit en général de maladies de survenue tardive qui touchent des tissus très différenciés de façon très sélective, comme des cardiomyopathies avec troubles de conduction, ou des maladies systémiques, comme des syndromes de vieillissement prématuré. Plusieurs de ces maladies ont été reproduits par inactivation génique de LMNA chez la souris.
Un des traits principaux de ces maladies est la perte musculaire que l’on observe dans la dystrophie musculaire d’Emery-Dreifuss, la cardiomyopathie dilatée associée aux anomalies de LMNA et la dystrophie musculaire des ceintures (limb-girdle muscular dystrophy).
Alors que les mécanismes physiopathologiques conduisant à ces phénotypes restent obscures, deux hypothèses de travail sont évaluiées actuellement : les anomalies de régulation génique et les anomalies de l’architecture nucléaire à l’origine d’une fragilité cellulaire. En effet, il y a un lien direct entre l’absence de protéine LMNA et la fragilité nucléaire des cellules vivantes. Cette anomalie nucléaire indique une perte d’interaction entre le noyau et le cytosquelette environnant. En effet l’organisation tridimensionnelle de filaments d’actine, de vimentine et de tubuline est anormale dans les cellules MEF-/-. Cette perte de la rigidité nucléaire est associée à une perte des interactions physiques entre les structures nucléaires, comme les lamines, et le cytosquelette.
La lame nucléaire (nuclear lamina) contribue à la forme, la taille et l’intégrité du noyau, le nombre et la position des pores nucléaires, ainsi que l’organisation de l’hétérochromatine.
Les lesions musculaires causées par les mutations de la lamine A/C sont associées èa une perte d’intégrité nucléaire après des traumatismes répétés, comme dans les maladies causées par les anomalies des filaments intermédiaires cytoplasmiques. De plus, à la différence des ces dernières, une part importante des mutations pathogènes de la lamine A/C sont situées en dehors de l’hélice, dans la queue de la lamine A/C. Cependant, la plupart des tissus affecté par les laminopathies ne sont pas exposées particulièrement aux traumatismes.
Pathogénie des laminopathies
La pathogénie des laminopathies est probablement associée aux autres fonctions des lamines encore peu connues, comme le contrôle de l’expression génique. Le rôle des lamines dans le contrôle de l’expression génique est suggéré par la liaison à la chromatine, à des facteurs de transcription spécifique, dont certains spécifiquement exprimés dans les adipocytes, et des protéines impliqués dans certains processus nucléaires comme la réplication de l’ADN ou la maturation des ARN.
Par ailleurs, les lamines exercent une action sur le réticulum endoplasmique, contigu à l’enveloppe nucléaire. En effet, des composants clés de l’enveloppe nucléaire comme l’émérine sont répartis de façon anormale dans les citernes du reticulum endoplasmique des fibroblastes de patients porteurs d’une mutation de la lamine A/C. Le lien entre ces mutations et les dysfonctions du reticulum endoplasmique ne sont pas encore connu : elle pourrait entrainer un stress cellulaire global ou altérer la réponse cellulaire aux stress spécifiques, comme le choc thermique.
D’autres études seront nécessaires pour connaître les effets variables et pléiotropes des mutations de la lamine. Ainsi, la structure atomique du domaine de queue de la lamine A montre que les mutations de la lamine A/C à l’origine des lipodystrophies partielles familiales et la dystrophie musculaire d’Emery-Dreifuss touche ce domaine de façon différente et pourrait agir en perturbant les interactions avec des protéines partenaires.
Pathologie des microfilaments d’actine
[modifier | modifier le wikicode]Les microfilaments, ou microfilaments d’actine, forment deux brins enroulés d’actine de 7 nm de diamêtre et peuvent mesurer jusqu’à plusieurs centimêtres de long. Ce composant du cytosquelette joue un rôle critique dans les changements de forme et la motilité de la cellule.
L’actine forme la composante principale des filaments fins des cellules musculaires et des microfilaments du cytsquelette des cellules non-musculaires. Les séquences d’acides aminés des différentes isoformes d’actine sont très homologues et trèes conservées au cours de l’évolution. Plusieurs classes d’actines sont distingués : les alpha-actines (gènes ACTA1 et ACTA2), beta-actins (ACTB), actines cardiaques (ACTC), les actines gamma (ACTG1, ACTG2). Les isoformes bêta et l’actine-gamma diffèrent par une substitution de 4 acides aminés à la partie N-terminale de la molécule. Ils sont co-exprimés dans les cellules non-musculaires.
Les fonctions du cytosquelette d’actine sont multiples : il maintient ou modifie la forme de la cellule, règle la division cellulaire, assure la transmission des signaux externes, maintient en place certaines organelles cellulaires. Il joue également un rôle clé dans la motilité cellulaire et dans le mouvement de vésicules cytoplasmiques à l’intérieur de la cellule. Il peut également jouer des rôles-clés dans des fonctions cellulaires spécifiques, comme l’activation des cellules T au cours de la réponse immune et la phagocytose.
Régulation des microfilaments d’actine
[modifier | modifier le wikicode]Plusieurs familles de protéines contrôlent la formation et la fonction des microfilaments d’actine : - les petites GTPases de la famille Rho comme Rho (MIM.165370), RAC (MIM.602048), et CDC42 (MIM.116952) - les 7 membres du complexe ARP2/3 complex (MIM.604221) - la protéine WASP (MIM.301000), mutée dans le syndrome de Wiskott-Aldrich, la protéine WASP-like (WASL) (MIM.605056), et WASF1.
WASP (MIM.301000), WASP-like (WASL) (MIM.605056) et WASF1 sont des transducteurs impliqués dans la transmission des signaux venant récepteurs tyrosine kinase et de petites GTPases vers les microfilaments d’actine.
La protéine WAVE est une protéine homologue de la verproline de la famille des WASPs qui forme avec WASP le réseau WASP-WAVE qui lie les signaux d’amont avec l’activation du complexe ARP2/3, qui induit la polymérisation rapide du microfilament d’actine. Cette polymérisation est cruciale pour la réorganisation du réseau cytosquelettique d’actine à la périphérie de la cellule lors de processus comme la motilité cellulaire, le trafic vésiculaire et aux cours des infections cellulaires. D’importantes familles de protéines liées à la membrane interagissent avec les protéines de la famille de WASP et de WAVE, apportant un deuxième niveau de régulation dépendant de la membrane à la polymérisation des microfilaments d’actine.
Pathologie
[modifier | modifier le wikicode]Les mutations des gènes codant les protéines d’actine sont à l’origine de cardiomyopathies dilatée et/ou hypertrophique, de myopathies de type némaline ou avec excès de myofilaments fins et d’une forme de surdité progressive dominante.
Les gènes de la famille Rho jouent un rôle dans les processus tumoraux. Les mutations germinales de la protéine WASP sont à l’origine du syndrome de Wiskott-Aldrich et de la thrombocytopénie liée à l’X (XLT) .
Par ailleurs, la polymérisation des microfilaments d’actine peut aussi être perturbée par les anomalies de la voie des inositols phosphates, comme dans la maladie de Lowe, due à des mutations du gène OCRL1, qui code la phosphatidylinositol 4,5 bisphosphate 5-phosphatase (#12428211#).
Cofilines
Les cofilines (CFLs: CFL1 et CFL2) sont des protéines ubiquitaires régulant l’actine. Elles lient et dépolymérisent les filaments d’actine F et inhibent la polymérisation de l’actine-G monomériques de façon pH dépendante. Elles sont impliquées dans la translocation du complexe actine-cofiline du cytoplasme vers le noyau. En pathologie, la voie des cofilines est un déterminant majeur des métastases. En effet, l’activité globale de la voie des cofilines (et non une protéine unique), détermine le phénotype invasif ou métastatique des cellules tumorales. Des inhibiteurs de la voie des cofilines sont actuellement en développement et pourrait avoir une action anti-métastatique.
Pathologie des microtubules
[modifier | modifier le wikicode]Les microtubules sont des éléments essentiels et ubiquitaires du cytosquelette qui jouent un rôle clé dans de nombreux processus cellulaires fondamentaux comme la division cellulaire, le transport intracellulaire et le maintien de l’architecture cellulaire.
Les microtubules sont des tubes formés de deux protéines appelées tubulines. Ils contrôlent le mouvement des cils et des flagelles, le mouvement des organelles dans le cytoplasme et le mouvement des chromosomes durant la mitose et la méiose. Les sous-unités à partir desquelles sont assemblées les hétérodimères de tubuline, sont la tubuline-alpha1 et la tubuline-beta1, chacun d’à peu près 50 kDaltons.
Leur fonction dépend de leur capacité à réarranger leur distribution à différentes périodes et dans différentes localisations. Les microtubules sont des polymères dynamiques et leur comportement peut être décrit comme une instabilité dynamique. Les microtubules jouent un rôle central dans la régulation de la forme cellulaire et la polarité au cours de la différentiation, la séparation des chromosomes au cours de la méiose et le transport intracellulaire. Les microtubules subissent des réarrangements qui supposent des transitions rapides entre des états stables et dynamiques pendant ces processus. Les microtubules contrôlent l’adaptation et la maintenance du cytosquelette. La polymérisation et la dépolymérisation des tubulines-alpha (MIM.602529) et bêta (MIM.191130) contrôlent l’assemblage et le désassemblage des microtubules. Les dysfonctions des cils cellulaires entraînent les dyskinésies ciliaires primitives (DNAH5, DNAH7, DNAH11), des anomalies de la latéralisation (situs inversus), des infertilité chez l’homme, où une association des trois dans le syndrome de Kartagener.
Pathologie des tubulines
Pathologie des protéines chaperones associées aux tubulines
Les protéines associées aux microtubules ou MAPs
Les protéines associées aux microtubules ou MAPs (MIM.600178) régulent la dynamique et la stabilité des microtubules, et les différents types de microtubules sont associés à différentes MAPs.
Les anomalies des MAPs entraînent une anomalie de la stabilité des microtubules. Les anomalies of the protein tau (TAU) sont à l’origine des taupathies. Les MAPs se lient aux microtubules et conditionnent leur stabilité et leurs interactions avec les autres composantes cellulaires. Au cours de la neurogenèse, par exemple, l’assemblage des microtubules constituent une étape essentielle, régulée par les MAPs. Les MAPs se divisent en deux groupes principaux selon leur masse moléculaire. Les MAPs de haut poids moléculaire comprennent MAP1A, MAP1B (MIM.157129), et MAP2 (MIM.157130) et un groupe de poids moléculaire moyen, qui comporte l’abondante protéine tau (TAU). MAP1B, également appelé MAP5, est un des composants de ponts entre microtubules. Il s’agit d’une molécule filament avec une petite extrémité sphérique. Les anomalies de la protéine tau (ou Taupathies) Tau (MAPT) est une protéine associée aux microtubules (MAPs). Dans plusieurs maladies neurologiques, appelées taupathies, des altérations de la protéine Tau entrainent des dégénération neuronale dans des régions spécifiques. Ces ‘taupathies’ sont dues à des agrégations de la protéine Tau en polymères fibrillaires. Bien que les régions cérébrales atteintes diffèrent dans les différentes taupathies, ces maladies ont des caractéristiques communes : une hyperphosphorylation anormale de Tau et une agrégation aberrante de Tau. Des mutations de Tau sont observées dans des cas familiaux de démence frontotemporale. Ces mutations réduisent la capacité de Tau à promouvoir l’assemblage de microtubules. Les différentes mutations de tau entraînent une anomalie des interactions de la protéine tau avec les microtubules, entraînant une hyperphosphorylation de la protéine Tau, son assemblage en filaments et la mort cellulaire qui en découle. Des mutations germinales de MAPT sont observées dans la démence frontotemporale plus ou moins associée à un syndrome de Parkinson (MIM.600274), la dégénération pallidopontonigrale (PPND) (MIM.168610), la maladie de Pick. Par ailleurs, des agrégations de la protéine Tau sont observées dans plusieurs maladies (maladie d’Alzheimer, maladie de Pick, démence fronto-temporale (MIM.600274), dégénération corticobasale, paralysie progressive supranucléaire. Ces agrégations forment d’abondantes inclusions cytoplasmiques formées de protéine tau hyperphosphorylée.
Thérapeutique
Em thérapeutique anti-cancéreuse, les microtubules sont les cibles de plusieurs drogues anti-cancéreuses. Les composés chimiques qui interfèrent avec les microtubules sont les alcaloïdes du vinca et les taxanes, qui forment deux groupes de médicaments très importants pour le traitement des proliférations cancéreuses. Leur activité est plus lié à leurs effets inhibiteurs sur la dynamique de l’épine de microtubules (spindle microtubule), que sur les leurs effets sur la masse des microtubules polymères.
Même de petites altérations de la dynamique des microtubules peuvent engager le spindle checkpoint, arrêter la progression du cycle cellulaire en mitose et éventuellement conduire à l’apoptose cellulaire.
D. Dans certains types cellulaires
Cytosquelette érythrocytaire
Au moins 15 types protéiques sont impliqués dans la formation du squelette membranaire des érythrocytes humains. Les mutations de plusieurs de ces protéines, comme les spectrines (SPTs), les ankyrines (ANKs), la protéine ‘band 3’ (SLC4A1) et la protéine band 4.1 (EPB41), sont associées à une fragilité accrue de érythrocytes et leur lyse.
Des homologues de ces protéines membrane-cytosquelette érythrocytaire sont retrouvées dans de nombreux autres types cellulaires. Dans les neurones cérébraux et les jonctions neuromusculaires, on en retrouve dans les densités post-synaptiques, ce qui suggère leur importance dans le maintien de la forme cellulaire et de la stabilité membranaire.
Dans le muscle
La protéine mutée dans la dystrophie musculaire de Duchenne (DMD) et la myopathie de Becker, la dystrophine, est une protéine associée à l’actine. Il s’agit d’un composant essentiel de la membrane sarcolemmal, qui la lie aux filaments d’actine.
Dans les cellules immunitaires
Les voies de signalisation conduisant au remodelage du cytosquelette d’actine jouent un rôle crucial dans l’activation des lymphocytes et la mobilité des cellules présentatrices de l’antigène (APC), comme les cellules dendritiques. Lors de l’interaction entre la cellule présentatrice de l’antigène (APC), comme les cellules dendritiques, la voie de signalisation du récepteur T (TCR) conduit à des évènements précoces comme la phosphorylation du TCR, le relargage du calcium Ca2+, au recrutement et l’activation de tyrosine kinases non-récepteur et de protéines adaptateurs.
Le complexe Arp2/3 et les protéines Ena/VASP co-localisent dans la synapse immunologique, la structure d’interaction entre les lymphocytes T et l’APC. Ils permettent de remodeler localement le cytosquelette d’actine. Les fonctions du complexe Arp2/3 et des protéines Ena/VASP sonnt coordonnées par la formation de complexe multimoléculaire similaires au cours de la phagocytose médié par le récepteur Fc-gamma.
Cytosquelette et infections microbiennes
[modifier | modifier le wikicode]Le cytosquelette joue un role très important dans la pathogenèse microbienne. Par exemple, la bactérie intracellulaire Listeria monocytogenes a la capacité de ce déplacer dans le cytoplasme des cellules qu’elle envahit en exploitant les mécanismes moléculaires de base des filaments d’actine.
Pour cela, la bactérie recrute à sa surface des composants clés de cette structure comme la protéine d’actine, le complexe Arp2/3, les protéines Ena/VASP et le complexe profiline-actine réarrangée en une structure appelée ‘queue d’actine’.
La polymérisation de l’actine à l’interface entre la queue d’actine et la partie postérieure de la bactérie fournit la force propulsive qui pousse la bactérie dans le cytoplasme. Le complexe Arp2/3 est également essentiel pour ce déplacement.
Septines
[modifier | modifier le wikicode]Les septines sont des éléments protéiques du cytosquelette.
Les septines sont impliquées dans la formation du cytosquelette, la division cellulaire et la tumorigenèse.
Les septines constituent une famille de protéines conservées qui forme des complexes hétéro-oligomériques qui s’assemblent en filaments. Ces filaments peuvent s’organiser en rangées linéaires, en bobines, en anneaux ou en mailles.
Les septines servent de structures paramembranaires et de barrières pour délimiter des compartiments locaux, en particulier pour établir un plan de clivage lors de la cytokinèse ou cytodiérèse, la division de la cellule lors de la division cellulaire (mitose).
La formation des complexes multi-septines est régulée par des nombreuses interactions protéine-protéine. La liaison du GTP et la phosphorylation dirigent la polymérisation des filaments requise pour l’assemblage des complexes de septine.
Cette famille regroupe 9 protéines numérotées de 1 à 9 (SEPT1 à SEPT9). En pathologie, des mutations germinales de SEPT9 sont observées dans l’amyotrophie neuralgique héréditaire (HNA).
Par ailleurs, le gène SEPT5 est un partenaire du gène MLL pour former un gène de fusion SEPT5/MLL (PNUTL1/MLL) par translocation t(11;22)(q23;q11.2) dans certaines leucémies myéloïdes aiguës.